






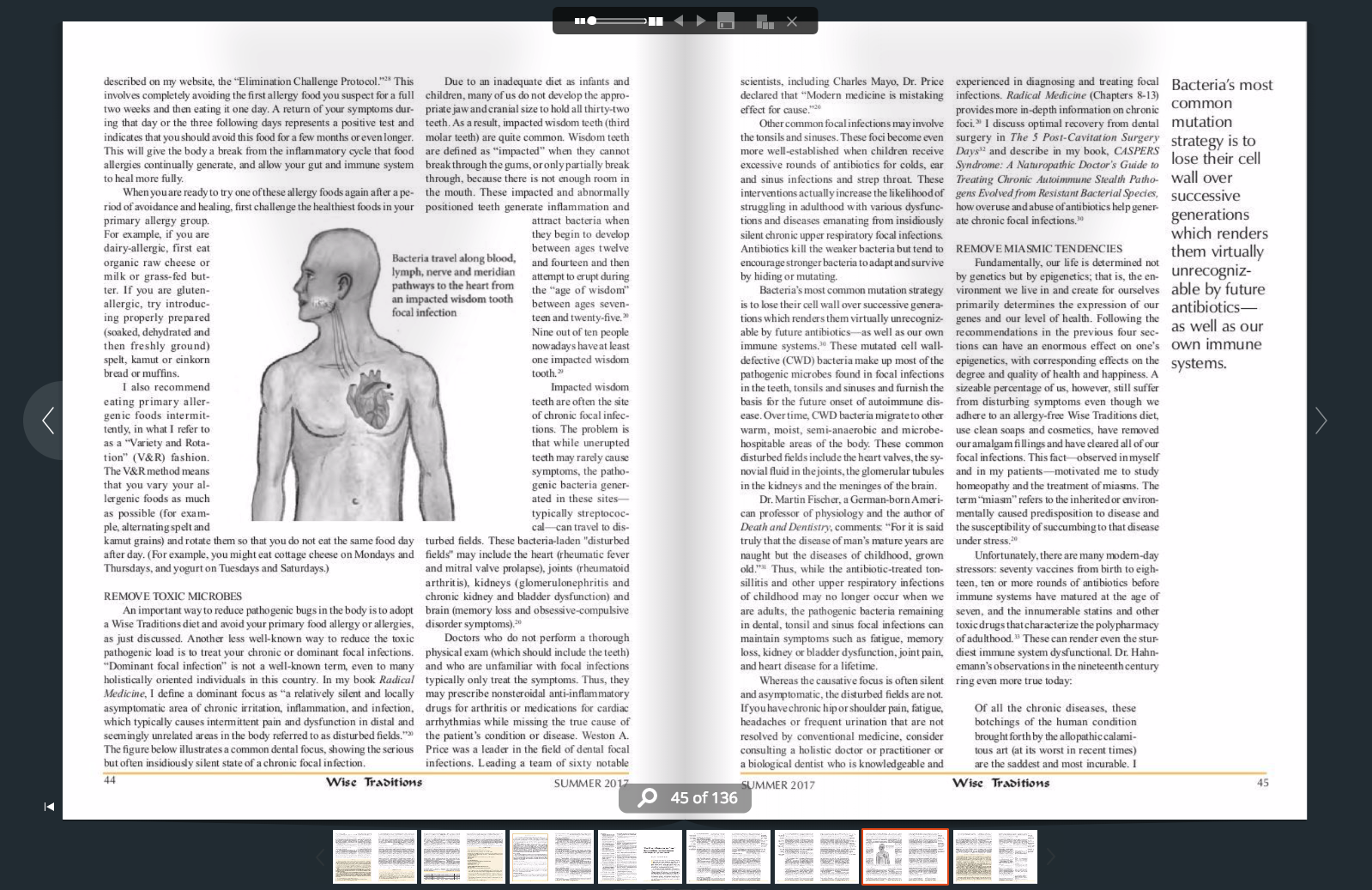
 🖨️ Print post
🖨️ Print post
ARTICLE SUMMARY
- Impaired sulfate supply to the heart is a key factor in cardiovascular disease.
- Red blood cells, platelets and cells in the skin synthesize cholesterol sulfate catalyzed by sunlight.
- Cholesterol sulfate, unlike cholesterol, is water soluble, so it can travel freely in the blood rather than packaged up inside an LDL particle.
- Glyphosate, the active ingredient in the pervasive herbicide Roundup, disrupts sulfate synthesis in the skin and disrupts bile flow from the liver, leading to a systemic deficiency in cholesterol sulfate.
- Sulfate provides negative charge in the blood vessel wall and for the red blood cells and platelets, promoting flow.
- Sulfate also maintains the structured water that lines the vessel walls and presents a slick, frictionless surface to the red blood cells.
- The atheroma actively recruits cholesterol to be ready to produce cholesterol sulfate when sulfate becomes available.
- Inflammation, while damaging to surrounding tissues, performs a useful service by promoting an oxidative environment necessary to make sulfate.
- A heart attack is a well-choreographed sequence of events aimed to restore sulfate supplies by oxidizing taurine, which is stored in large amounts in the heart.
- Statin drugs, by reducing the supply of cholesterol sulfate to the heart, will lead to heart failure down the road, a worse prognosis than cardiovascular disease.
Despite decades of research, atherosclerosis remains a poorly explained phenomenon. The simple story experts present to the public is that excess cholesterol accumulates in the blood and combines with other substances to form an atheroma (also called plaque) that lodges in the artery wall, eventually obstructing flow. Strikingly, however, the lipid deposits accumulate only in arteries and never in veins. Even more striking is the fact that the arteries supplying the heart are the most vulnerable.
The conventional explanation leaves unanswered many questions about the pathogenesis of atherosclerosis and cardiovascular disease. Why does cardiovascular plaque accumulate only in arteries, and preferentially in arteries supplying the heart? What prompts the occurrence of inflammation, which many believe to be a critical factor in heart disease? Why have studies on vitamin D supplementation proved disappointing despite research showing a strong inverse relationship between sunlight availability and heart disease?
To answer these questions, I propose a unifying theory for the etiology of cardiovascular disease. The theory involves cholesterol sulfate, a molecule that circulates in the bloodstream and performs a variety of important regulatory functions. I believe that the cause of heart disease is an inadequate supply of cholesterol sulfate to the heart. When there are pathologies that impair the normal conditions for making cholesterol sulfate, an atheroma develops as an alternative means of supplying the heart with vital cholesterol sulfate. When neither the normal nor back-up mechanisms are able to make adequate cholesterol sulfate, the outcome is heart failure, a much worse prognosis than atherosclerosis.
A number of diverse observations involving various forms of sulfur support the hypothesis that impaired sulfur supply to the vasculature is the key factor in cardiovascular disease. For example, early studies on primates showed that a high-fat, high-cholesterol diet fed to monkeys (not a normal diet for them!) could induce atherosclerosis, but that simultaneous supplementation with sulfur-containing nutrients was protective.1 Similarly, experiments on rats showed that a diet supplemented with excess cholesterol, cholic acid (a bile acid) and vitamin D2 could induce aortic lesions expressing calcification and plaque formation, but it was possible to prevent such lesions completely through simultaneous supplementation with chondroitin sulfate (a modified sugar molecule containing oxidized sulfur).2
As another example, children with disorders in the metabolism of cysteine (a sulfur-containing amino acid) develop atherosclerosis-like arterial damage at an early age.3 Interestingly, the consumption of garlic—a rich source of sulfane sulfur—is inversely correlated with the progression of cardiovascular disease.4 Finally and remarkably, synthetic hydrogen sulfide donors (molecules that release therapeutic hydrogen sulfide) can protect mitochondria in endothelial cells (the thin layer of cells that line the interior of blood vessels) from oxidative damage.5
CHOLESTEROL SULFATE SYNTHESIS
Normally, keratinocytes (the predominant cells found in the epidermis), red blood cells (RBCs) and platelets produce cholesterol sulfate in the skin, catalyzed by sunlight. Therefore, in accordance with my unifying theory, one would expect sunny climates to reduce heart disease risk. Indeed, geographical data show an inverse relationship between cardiovascular disease and annual sunlight availability.6 France and Spain have much lower rates of death from heart attacks than the United Kingdom. In a study conducted in the British Isles, mean annual sunshine hours accounted for 49 percent of the variance in mortality from coronary heart disease.7
Despite related studies showing that vitamin D deficiency is associated with cardiovascular disease risk,8 placebo-controlled trials have failed to demonstrate any benefit from vitamin D3 supplementation.9 Why? My coauthors and I suggest that, where heart disease is concerned, the benefit of sunlight comes from cholesterol sulfate synthesis rather than vitamin D3 synthesis. In a 2012 article in Entropy,10 we proposed that sulfate is produced from reduced sulfur sources in the skin, catalyzed by sunlight.
The enzyme that likely carries out this function is endothelial nitric oxide synthase (eNOS), the same enzyme that produces nitric oxide to relax the artery wall. The enzyme eNOS is a member of the cytochrome P450 (CYP) superfamily of enzymes, which metabolize drugs and synthesize cholesterol, steroids and other lipids. I hypothesize that the overuse of sunscreen has played a dual damaging role, suppressing sunlight catalysis but also actively disrupting eNOS’s sulfate-producing function due to sunscreen’s aluminum content.11 Many other environmental chemicals also disrupt CYP enzymes, including mercury12,13 arsenic,14 cadmium12 and glyphosate,15,16 the active ingredient in the pervasive herbicide Roundup. When aluminum or other toxic chemicals disable eNOS, it is unable to make enough sulfate to supply the needs of the endothelial cells, creating a systemic sulfate deficiency problem.
It is well established that eNOS produces superoxide as well as nitric oxide, but biologists have always viewed this as a pathology.17 At the same time, the fact that RBCs contain abundant eNOS baffles biologists, because nitric oxide would disrupt hemoglobin’s ability to transport oxygen.18 Viewing eNOS superoxide synthesis as an alternative function to oxidize sulfur offers an explanation for both of these puzzles.
GLYPHOSATE AND HYPERLIPIDEMIA
Getting back to coronary artery disease, most people assume that elevated serum lipids (hyperlipidemia) are a causal factor. Tens (if not hundreds) of millions of people receive advice from their doctor to take a statin drug to protect them from heart disease because their serum lipid levels are high. Therefore, one would expect a plot over time of the hospital discharge rates for hyperlipidemia to be highly correlated with a similar plot for coronary artery disease. This is not the case, however. As the figure below shows, the correlation coefficient value is a weak 0.39 (whereas greater than .70 would indicate a strong correlation), with an insignificant p-value of 0.19. Although correlation does not always imply causation, it is surprising and noteworthy when a factor presumed to be causative is not correlated with the disease.
In comparison, there is a very strong and highly significant correlation between hyperlipidemia and glyphosate application to corn and soy crops (R = 0.97, p < .000018), as shown in the second figure. Glyphosate application rates have steadily increased over time due to the widespread appearance of glyphosate-resistant weeds growing among the crops that are increasingly engineered to be Roundup Ready. Serum lipids have risen in tandem with the increase in glyphosate use, despite the increase in statin drug prescriptions. As the second figure suggests, it is not unreasonable to propose that glyphosate is causal in hyperlipidemia.
Glyphosate disrupts CYP enzyme activity in the liver, which can explain the contribution of glyphosate to hyperlipidemia.19 A rat study assessing the impact of glyphosate, clofibrate (a cholesterol-lowering drug) and two phenoxyacid herbicides on liver function showed that glyphosate reduced the activity of CYP enzymes in the liver much more than the other substances investigated.19 Multiple CYP enzymes are needed to produce bile acids,20 which facilitate digestion and absorption of lipids and regulate cholesterol homeostasis. Bile acids normally export a large amount of cholesterol via the digestive system. Impeded bile acid synthesis due to a defective CYP7A1 gene produced neonatal cholestasis (blocked bile ducts) and hypercholesterolemia (specifically, elevation in serum LDL) in mice fed a normal chow diet.21
Given that eNOS is a CYP enzyme, it is entirely plausible that glyphosate disrupts eNOS’s ability to synthesize sulfate. We would expect eNOS exposure to glyphosate in the red blood cells, because glyphosate export via the kidney requires transit through the vasculature. Disrupted synthesis of cholesterol sulfate will necessitate an increase in the synthesis of LDL particles to transport cholesterol in its unsulfated form, because cholesterol is not water-soluble and therefore must be stored inside a lipid particle for transport.
ATHEROMA AS ALTERNATIVE SUPPLIER OF CHOLESTEROL SULFATE
In another Entropy publication, my coauthor and I suggested that when red blood cells produce cholesterol sulfate while traversing the surface veins, catalyzed by sunlight, they release the cholesterol sulfate to the tissues in the capillaries.22 This refurbishes both the cholesterol and the sulfate supply to the endothelial wall, maintaining vascular health.
The glycocalyx is a complex mesh of sulfated sugar chains that lines the interior wall of all blood vessels and is important for vascular health. Heparan sulfate proteoglycans (HSPGs) in the glycocalyx play many important roles,23 mediating cellular signaling mechanisms and promoting uptake of various nutrients, including LDL clearance by liver cells.24 Perhaps the most important role of cholesterol sulfate in the capillaries is to maintain a thick layer of gelled water coating the inner surface. This structuring effect on water is due to the “kosmotropic” properties of sulfate,25 which provides a near-frictionless surface contact with the red blood cells10 that is further enhanced by the negative charge in both the glycocalyx and the red blood cell membrane.
As the red blood cells drop off cholesterol sulfate in the capillaries, they also lose their negative charge, which means that the venous end has a lower pH than the arterial end. An increase in carbon dioxide content on the venous side enhances this effect, as is immediately following cardiac arrest, when carbon dioxide accumulates in the veins and the voltage difference between arteries and veins sharply increases.26 Thus, an electrical gradient also propels the negatively charged red blood cells through the capillaries. Capillary resistance is the dominant factor in high blood pressure, which we can therefore expect to correlate with an impoverishment in cholesterol sulfate in red blood cell membranes and heparan sulfate in the capillary walls. Both increased friction at the walls and decreased force from the electromagnetic field impede movement of red blood cells through the capillaries.
The glycocalyx is constantly shed and rebuilt in a dynamic process that is promoted by inflammatory agents.27 Complement and endotoxin (from bacteria) both induce glycocalyx shedding through a G-protein coupled receptor response. Such matrix remodeling is especially active during ischemia and reperfusion (the two critical stages of a heart attack). Membrane-bound matrix metalloproteinases (MMPs) can detach fragments of the glycocalyx from the artery wall, which can then be redistributed to other parts of the vasculature (such as the capillaries) as reinforcements. Thus, it is highly conceivable that the glycocalyx in atherosclerotic regions is a source of raw materials needed to maintain the health of the heparan sulfate proteoglycans (HSPGs) in the capillary glycocalyx.
As an alternative means of supplying cholesterol sulfate to the heart, an atheroma is uniquely suited to cholesterol sulfate’s manufacture by platelets. The sulfate is supplied by breaking down homocysteine thiolactone, a precursor that is ready to become sulfate under the right circumstances; the lipid stores in the macrophages supply the cholesterol; the red blood cells supply adenosine triphosphate (ATP) to energize the reaction; and the inflammatory response provides superoxide needed to oxidize the sulfur atom in homocysteine.10 HDL-cholesterol plays a critical role, because platelets will take up cholesterol only from AI-HDL (the “good” variant of HDL), and they will increase their production of cholesterol sulfate three-hundred-fold in the presence of 3’-phosphoadenosine-5’-phosphosulfate (PAPS), a source of transferrable sulfate produced by sulfation of ATP.28 (Note that when ATP is in short supply, this will affect the ability to utilize any sulfate that is synthesized, resulting in blood with overly high viscosity due to sulfate’s property of gelling the blood.)
The lipoprotein apolipoprotein E (ApoE) plays a significant role in inducing the export of cholesterol from the macrophages into AI-HDL, but it also induces enrichment of sulfate in the HSPGs.29 This implies that it escorts cholesterol sulfate rather than cholesterol in its unsulfated form out of the cell. This idea is also supported by the fact that cholesterol sulfate, unlike cholesterol, is water-soluble, and therefore readily crosses the cytoplasmic gap between the endothelial reticulum and the plasma membrane. Both homocysteine30 and gamma-glutamyltransferase (GGT)31,32 are risk factors for heart disease, and both can be explained because they provide substrate for sulfate synthesis when the normal sun-catalyzed sulfate synthesis mechanisms are not working. GGT breaks glutathione down into cysteinylglycine and glutamate, and cysteine from cysteinylglycine can be oxidized to form sulfate.33
Atheromata (plural of atheroma) harbor various pathogenic microbial species, the most significant of which is probably Chlamydia pneumoniae.34,35 The strong association between chronic C. pneumoniae infection and atherosclerosis has prompted some researchers to identify it as the pathogen implied in a chronic infection theory of heart disease. While many proponents of this hypothesis enthusiastically have embraced the concept of antibiotic treatment specific to C. pneumoniae, clinical trials have been disappointing.36
C. pneumoniae are dormant except when internalized into host cells. Within these cells, they produce a unique form of heparan sulfate, using a set of enzymes that are not found in any other known bacterial species.37 I hypothesize that C. pneumoniae play a special role in enhancing the supply of heparan sulfate to an atheroma, and they may well be able to do so in the absence of functional CYP enzymes.
MULTIPLE PATHWAYS TO HEART FAILURE
Deaths from heart attack have steadily decreased in the industrialized world over the past few decades,38,39 but we are simultaneously experiencing an increasing health care burden in an emerging epidemic of heart failure.40 In fact, heart failure is the single most frequent cause of hospitalization for those over sixty-five years old, affecting five million Americans as of 2010.41 I maintain that heart failure is a direct sequela to insufficient supply of cholesterol and sulfate to the heart. Since an atheroma plays a significant role in supplying these nutrients, it can be anticipated that factors that interfere with the healthy function of the atheroma will lead, over time, to heart failure.
Other pathways to heart failure furnish additional observations. For example, Chagas disease is an infectious disease endemic to regions of South America caused by the Tryptanosoma cruzi pathogen.42,43 Patients who recover are susceptible to premature death by heart failure many decades later. These individuals suffer from frequent small heart attacks but are remarkably free of atherosclerosis.43 The explanation for this unique profile follows logically from the fact that T. cruzi produces an antigenic molecule that closely mimics cholesterol sulfate.42 As a result, Chaga disease patients develop antibodies to cholesterol sulfate. This would render the synthesis of cholesterol sulfate by an atheroma counterproductive while leading to system-wide deficiencies in cholesterol and sulfate.
Down syndrome (trisomy 21) provides another interesting example. Down syndrome is associated with a low risk of atherosclerosis44,45 combined with premature susceptibility to heart failure46 and Alzheimer’s disease.47 A key enzyme present on chromosome 21 is Cu/Zn superoxide dismutase (SOD), which is 50 percent overexpressed in association with Down syndrome.48 SOD dismutates superoxide to hydrogen peroxide, thus reducing the bioavailability of superoxide for the oxidation of reduced sulfur sources (such as homocysteine thiolactone and cysteinylglycine derived from glutathione)31 to form sulfate. Down syndrome patients are susceptible to Alzheimer’s at least twenty to thirty years earlier than one would normally expect, and dementia is clinically detected in association with Down syndrome at least three times more frequently than in individuals without trisomy 21.49 It is probable that sulfate deficiency plays a significant role in the Alzheimer’s brain, as evidenced by the severe deficiency in sulfatide, the only sulfonated lipid, observed in association with Alzheimer’s disease.50
Statin drugs likely represent another pathway to heart failure. Many have argued that statin drugs produce pleiotropic effects (so-called beneficial effects that are not the drugs’ main or intended action) through an anti-inflammatory effect,51-53 and, further, that other treatments aimed at reducing inflammatory signaling might be effective treatments.54 I predict that both statin therapy and these other treatments instead will lead to heart failure. Statins disrupt G-protein coupled receptor signaling via their suppression of the synthesis of geranylgeranyl pyrophosphate, leading to defective protein prenylation (lipid modification),55 which explains their induction of arterial calcification.56 It is plausible that statin drugs promote heart failure through impairment of cholesterol sulfate synthesis in the atheroma, both by reducing the bioavailability of LDL to the atheroma and by interfering with the inflammatory response.
Not surprisingly, low cholesterol is consistently associated with poor survival statistics in heart failure.57-59 Total cholesterol levels under two hundred milligrams per deciliter increase the risk of dying from heart failure by up to three-fold.57,58 One group of researchers provocatively asks: “Could elevated total cholesterol, which is so firmly established to be deleterious for the development of coronary heart disease and coronary heart disease mortality, actually turn out to be good for patients with chronic [heart failure]?”59
THE ROLE OF TAURINE
Taurine plays a critical though as yet poorly understood role in cardiovascular disease. Taurine is the only sulfonated amino acid. The highest concentrations of taurine are found in the heart, where taurine represents 50 percent of the free amino acid pool,60 with large concentrations also stored in the brain and liver. Although some bacteria can utilize taurine as a fuel source,61 it has long been maintained that mammalian cells cannot metabolize taurine.62 However, preoperative infusion of taurine decreases reperfusion injury following coronary bypass surgery,63 and in rat studies, taurine supplementation has improved heart function following a heart attack.64
Blood platelets also retain taurine. Studies on dogs, cats and humans have demonstrated a direct linear relationship between plasma taurine levels and platelet taurine levels.65,66 Platelets from taurine-deficient cats and humans are more sensitive to clotting stimuli.66 Studies in dogs have shown that ischemia (heart attack) induced a 47 percent loss of taurine in the heart’s left ventricle and a 26 percent loss in both atria.67,68
Human white blood cells produce an enzyme called myeloperoxidase (MPO) during ischemia and reperfusion injury.69 Elevated MPO is an established risk factor for cardiovascular mortality following artery grafts.70 MPO often serves as a potent bacteriocidal weapon,71 potentially linking it to the infectious theory of heart disease.35,72 Hypochlorite, produced in response to MPO, avidly oxidizes many sulfhydryl-dependent proteins, leading to platelet activation and causing white blood cells to attach to the walls of blood vessels. It has been proposed that taurine protects from these damaging effects by reacting with and neutralizing hypochlorite to form taurine chloramine,73 a much more reactive compound than taurine. As we have seen, taurine is released from the heart during ischemia and would be readily available in the serum because it is not taken up by the platelets.74 However, taurine chloramine activates complement,75 which in turn promotes an inflammatory response, providing oxidizing agents that could in theory promote the oxidation of sulfur in taurine chloramine.
Thus, a novel way to view taurine is as another potential buffer for sulfate renewal in times of acute deficiency. Assuming that sulfate insufficiency is an important factor in heart disease, one should ask whether taurine chloramine can become a substrate for sulfate synthesis. Taurine chloramine already contains sulfur at an oxidation state of +5, so it only needs to oxidize the sulfur from +5 to +6 to produce sulfate. (A positive oxidation state shows the total number of electrons that have been removed from an element to get to its present state.)
Notwithstanding the claim that taurine is not easily broken down by mammalian cells, 25 percent of the traced sulfur in supplemental taurine turns up in the urine as sulfate.62 One hypothesis is that the gut bacteria metabolize the taurine to sulfate. Moreover, because taurine chloramine is more reactive, perhaps it can be metabolized to yield free sulfate, with microbial infective agents playing a facilitative role. If so, this would justify both cardiac storage of taurine as a back-up source of sulfate during times of severe deficiency as well as the presence of microbes in cardiovascular lesions,35,72 lending further credibility to the infectious theory of heart disease.
Taurine’s protection during ischemia and reperfusion occurs through an as yet undetermined mechanism.76 There is a severe loss of taurine from the heart during ischemia that can lead to heart failure if the taurine is not replenished. A World Health Organization population study revealed an inverse association between taurine excretion and ischemic heart disease mortality.77
![]()
Reperfusion causes a burst in oxygen consumption through diversion of the electrons from the electron chain into superoxide production.78,79 (The addition of one electron to oxygen produces superoxide.) Although superoxide—a reactive oxygen species—is generally viewed as “bad,” I argue that a key purpose of superoxide production in this instance is to oxidize sulfur (derived from serum cysteine or homocysteine reserves) from a negative oxidation state (-2) to a positive oxidation state (+6), producing sulfate from sulfide and consuming two superoxide anions. In other words, superoxide is necessary as a reactant to be able to produce sulfate from these reduced sulfur sources. Several enzymes working in conjunction with the mitochondrial electron chain are involved in oxidizing hydrogen sulfide to sulfate and thiosulfate.80 (In fact, thiosulfate may be the primary source of sulfur that is oxidized by eNOS.10) Homocysteine thiolactone is converted to sulfate in the presence of superoxide, catalyzed by vitamin A and vitamin C,81 possibly explaining the positive role of vitamin C in protection from cardiovascular disease.82
Taurine’s widespread health benefits to the cardiovascular system83 include its ability to inhibit the osteoblastic differentiation of vascular smooth muscle cells that leads to artery calcification.84 Taurine showed protection against the loss of mechanical function in rat hearts in both a heart failure and ischemia model.85 Taurine also suppresses the production of superoxide during reperfusion.86 I propose that this beneficial suppressive effect is achieved mainly because taurine already contains sulfur at a positive oxidation state of +5; hence only one-sixth as much superoxide is needed to produce an equivalent amount of sulfate (+6). The fact that taurine is normally inert also makes it a great choice for buffering as a precursor to sulfate. As previously noted, hypochlorite is needed to convert taurine into the more reactive molecule, taurine chloramine, from which sulfate can be derived with the help of superoxide and perhaps microbial enzymes. Species of clostridium are able to utilize sulfonate forms of sulfur such as taurine as an energy source. More generally, anaerobic bacteria can convert taurine directly to thiosulfate.61
Experiments have confirmed that taurine can be broken down to sulfoacetaldehyde by neutrophils, with taurine chloramine as an intermediary, through nonenzymatic hydrolysis catalyzed by hydrogen peroxide.87 With an adequate source of energy, sulfoacetaldehyde can react with phosphate to produce acetyl phosphate and sulfite. Sulfite can then be oxidized to sulfate via enzymatic action of sulfite oxidase. I propose that this reaction takes place during a heart attack. A China-based study of possible associations between biochemical, dietary and lifestyle factors and cardiovascular disease that did not find any relationship with serum cholesterol but showed a protective effect for molybdenum88 could be explained by the fact that molybdenum is a cofactor for sulfite oxidase.
PREVENTION AND TREATMENT OF CARDIOVASCULAR DISEASE
If the ideas proposed here are valid, they suggest some very simple measures that can be taken to decrease the risk of cardiovascular disease. Consuming a strictly organic diet that is rich in sulfur-containing foods and spending significant time outdoors without sunscreen on sunny days (exposing both the skin and the eyes to the sun) are two important lifestyle changes. Conversely, the unifying theory suggests that sun avoidance and consumption of chemical- and glyphosate-laden processed foods are likely to be major contributors to the development of coronary atherosclerosis.
One would expect sulfur-rich soil and water derived from basalt rock to be cardioprotective. People living on islands enriched with sulfur-containing volcanic basalt rock (such as Japan, Iceland and Crete) enjoy a low risk of heart attacks as well as extended life expectancy. For those not living in such locations, many sulfur-containing compounds have been shown to benefit cardiovascular health, and a broad range of biologically active molecules—recognized as being cardioprotective—have in common that they supply sulfur to the body (Table 1, page 23).
THE BODY ELECTRIC
The topic of electricity in the body is beyond the scope of this article, but I want to leave you with an image of a solar-powered electrical circuit connecting all parts of the body, where the “wires” are the blood vessels. As noted, the biosulfates in the glycocalyx play an essential role in maintaining an “exclusion zone” of gelled water lining all the vessel walls, within which electrons are mobilized in the gel and protons at the interface to produce electrical current that powers the muscles and neurons.96-98 Thus, cholesterol sulfate captures sunlight energy in the bound sulfate anion to fuel both mobility and neuronal signaling, just as chlorophyll endows plants with the ability to convert sunlight energy into stored sugars, starches and fats. Life on the earth’s surface has always had access to sunlight as an energy source, and animals and plants have found distinct ways to utilize it.
Animals use sunlight to oxidize oxygen to superoxide which then oxidizes sulfur to sulfate. Cholesterol is the carrier molecule that distributes the sulfate over the vasculature. Cholesterol, too, is oxidized by sunlight to form vitamin D, a signaling molecule that communicates to the tissues that “all is well.” As the negatively charged red blood cells traverse the capillaries, they create a dynamic electromagnetic signal called the “streaming potential” that oscillates with the rhythm of the heartbeat. Endothelial cells respond to this signal by releasing nitric oxide, which relaxes the vessels and promotes flow.99
Cholesterol sulfate is an important source of sulfate to maintain the red blood cells’ negative surface charge and to populate the extracellular matrix of the endothelial wall. This results in near frictionless trafficking of red blood cells through capillaries.
CONCLUSION
Although the processes that take place in atherosclerotic regions of major arteries in the heart are complex, they can easily be explained as a mechanism to assure that the heart receives desperately needed cholesterol sulfate supplies. If eNOS were working properly to produce cholesterol sulfate, and the sulfate carriers that are supposed to be produced in the gut were working properly (derived from the aromatic amino acids coming out of the shikimate pathway that glyphosate disrupts), sulfate would already be well supplied and there would be no need for inflammatory back-up mechanisms to make sulfate “on the spot.”
I maintain that the modern lifestyle of sun avoidance and exposure to toxic chemicals through food, sunscreen or other environmental insults results in impaired cholesterol sulfate synthesis in the skin mediated by sunlight. The resulting pathology—severe sulfate deficiency—necessitates an alternative last-ditch mechanism for cholesterol sulfate synthesis. Thus, cholesterol gets stored in the artery wall to be made readily available for cholesterol sulfate synthesis whenever precursor sulfur sources such as homocysteine or cysteinylglycine are available, along with superoxide and ATP as sources of oxygen and energy to fuel the reaction. In an emergency, a heart attack also can initiate a programmed response that depletes taurine reserves to restore sulfate supplies. In addition, microbes such as C. pneumoniae can assist in replenishing heparan sulfate to the artery wall. It is no surprise that as statin therapy has interfered with even these less-optimal forms of cholesterol sulfate production, the risk of heart failure has increased, becoming a major contributor to hospitalization, rising health care costs and mortality.
SIDEBARS
PATHOLOGY OR THERAPY?
SUPEROXIDE/INFLAMMATION: Inflammation is necessary to trigger the cascade response that ultimately restores sulfate supplies to the vasculature through oxidation of sulfur-containing molecules like cysteine, homocysteine, hydrogen sulfide gas and taurine.
ATHEROMA: The atheromata in the artery wall supplying the heart actively recruit cholesterol and fats so that they can be ready to produce cholesterol sulfate once sulfate becomes available, and replenish the heart’s supply of this critical nutrient.
CLOSTRIDIA PNEUMONIAE: This microbe infects the artery wall where it can perform an important service, providing heparan sulfate to the heart.
HEART ATTACK: The complex cascade that takes place during a heart attack appears to be aimed at producing sulfate by oxidizing taurine molecules that are stored in the highest concentration in the heart and released during a heart attack.
WHY THE ATHEROMATA ARE CONCENTRATED IN CORONARY ARTERIES
The heart is arguably the most important organ in the body, and it needs a constant supply of cholesterol and sulfate in order to stay healthy and maintain its blood supply. Cholesterol is actively recruited into the arteries rather than the veins, because it can then be released as cholesterol sulfate once sulfate becomes available, which will then immediately enter the coronary capillaries to supply this critical nutrient to the heart’s vascular supply.
HOW TO PROTECT YOURSELF FROM HEART DISEASE
• Make sure that the foods you buy are certified organic, or know your farmer and verify that he or she does not use
pesticides.
• Eat sulfur-containing foods such as seafood, eggs, cheese, grass-fed beef, cruciferous vegetables, garlic and onions.
• Include red meat and fish in the diet; they are excellent sources of taurine.
• Get plenty of sun exposure to the skin and eyes without sunscreen and without sunglasses.
• Take frequent hot baths with Epsom salts added to the water.
• Just say “no” to statin drugs.
REFERENCES
1. Mann GV. Effects of sulfur compounds on hypercholesteremia and growth in cysteine-deficient monkeys. Am J Clin Nutr 1960;8:491-8.
2. Morrison LM, Bajwa GS, Alfin-Slater RB, Ershoff BH. Prevention of vascular lesions by chondroitin sulfate A in the coronary artery and aorta of rats induced by a hypervitaminosis D, cholesterol-containing diet. Atherosclerosis 1972;16(1):105-18.
3. McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 1969;56(1):111-28.
4. Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, et al. Hydrogen sulfide mediates the vasoactivity of garlic. Proc Natl Acad Sci U S A 2007;104(46):17977-82.
5. Szczesny B, Módis K, Yanagi K, Coletta C, Le Trionnaire S, Perry A, et al. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 2014;41:120-30.
6. Grimes DS, Hindle E, Dyer T. Sunlight, cholesterol and coronary heart disease. QJM 1996;89(8):579-89.
7. MacPherson A, Bacsó J. Relationship of hair calcium concentration to incidence of coronary heart disease. Sci Total Environ 2000;255(1-3):11-19.
8. Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008;117:503-11.
9. Wood AD, Secombes KR, Thies F, Aucott L, Black AJ, Mavroeidi A, et al. Vitamin D3 supplementation has no effect on conventional cardiovascular risk factors: a parallel-group, double-blind, placebo-controlled RCT. J Clin Endocrinol Metab 2012;97(10):3557-68.
10. Seneff S, Lauritzen A, Davidson R, Lentz-Marino L. Is endothelial nitric oxide synthase a moonlighting protein whose day job is cholesterol sulfate synthesis? Implications for cholesterol transport, diabetes and cardiovascular disease. Entropy 2012;14(12):2492-2530.
11. Tian Q-Y, Sun D-H, Zhao M-G, Zhang W-H. Inhibition of nitric oxide synthase (NOS) underlies aluminum-induced inhibition of root elongation in Hibiscus moscheutos. New Phytol 2007;174(2):322-31.
12. Alexidis AN, Rekka EA, Kourounakis PN. Influence of mercury and cadmium intoxication on hepatic microsomal CYP2E and CYP3A subfamilies. Res Commun Mol Pathol Pharmacol 1994;85(1):67-72.
13. Korashy HM, El-Kadi AOS. Modulation of TCDD-mediated induction of cytochrome P450 1A1 by mercury, lead and copper in human HepG2 cell line. Toxicology in Vitro 2008;22(1):154-8.
14. Anwar-Mohamed A, El-Sherbeni AA, Kim SH, Althurwi HN, Zordoky BN, El-Kadi AO. Acute arsenic toxicity alters cytochrome P450 and soluble epoxide hydrolase and their associated arachidonic acid metabolism in C57Bl/6 mouse heart. Xenobiotica 2012;42(12):1235-47.
15. Samsel A, Seneff S. Glyphosate’s suppression of cytochrome P450 enzymes and amino acid biosynthesis by the gut microbiome: pathways to modern diseases. Entropy 2013;15(4):1416-1463.
16. Samsel A, Seneff S. Glyphosate, pathways to modern diseases II: celiac sprue and gluten intolerance. Interdiscip Toxicol 2013;6(4):159-84.
17. Verhaar MC, Westerweel PE, van Zonneveld AJ, Rabelink TJ. Free radical production by dysfunctional eNOS. Heart 2004;90(5):494-495.
18. Kleinbongard P, Schulz R, Rassaf T, Lauer T, Dejam A, Jax T, et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood 2006;107(7):2943-51.
19. Hietanen E, Linnainmaa K, Vainio H. Effects of phenoxyherbicides and glyphosate on the hepatic and intestinal biotransformation activities in the rat. Acta Pharmacol Toxicol (Copenh) 1983;53(2):103-12.
20. Lorbek G, Lewinska M, Rozman D. Cytochrome P450s in the synthesis of cholesterol and bile acids – from mouse models to human diseases. FEBS J 2012;279(9):1516-33.
21. Erickson SK, Lear SR, Deane S, Dubrac S, Huling SL, Nguyen L, et al. Hypercholesterolemia and changes in lipid and bile acid metabolism in male and female CYP7A1-deficient mice. J Lipid Res 2003;44:1001-9.
22. Davidson RM, Seneff S. The initial common pathway of inflammation, disease, and sudden death. Entropy 2012;14(8):1399-1442.
23. Tumova S, Woods A, Couchman JR. Heparan sulfate proteoglycans on the cell surface: versatile coordinators of cellular functions. Int J Biochem Cell Biol 2000;32(3):269-88.
24. MacArthur JM, Bishop JR, Stanford KI, Wang L, Bensadoun A, Witztum JL, et al. Liver heparan sulfate proteoglycans mediate clearance of triglyceride-rich lipoproteins independently of LDL receptor family members. J Clin Invest 2007;117(1):153-64.
25. Collins KD. Charge density-dependent strength of hydration and biological structure. Biophys J 1997;72(1):65-76.
26. Grundler W, Weil MH, Rackow EC. Arteriovenous carbon dioxide and pH gradients during cardiac arrest. Circulation 1986;74(5):1071-4.
27. Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol 2004;286(5):H1672-H1680.
28. Yanai H, Javitt NB, Higashi Y, Fuda H, Strott CA. Expression of cholesterol sulfotransferase (SULT2B1b) in human platelets. Circulation 2004;109(1):92-6.
29. Paka L, Kako Y, Obunike JC, Pillarisetti S. Apolipoprotein E containing high density lipoprotein stimulates endothelial production of heparan sulfate rich in biologically active heparin-like domains: a potential mechanism for the anti-atherogenic actions of vascular apolipoprotein E. J Biol Chem 1999;274:4816-23.
30. McCully KS. Chemical pathology of homocysteine. IV. Excitotoxicity, oxidative stress, endothelial dysfunction, and inflammation. Ann Clin Lab Sci 2009;39(3):219-32.
31. Emdin M, Pompella A, Paolicchi A. Gamma-glutamyltransferase, atherosclerosis, and cardiovascular disease: triggering oxidative stress within the plaque. Circulation 2005;112(14):2078-80.
32. Mao Y, Qi X, Xu W, Song H, Xu M, Ma W, et al. Serum gamma-glutamyl transferase: a novel biomarker for coronary artery disease. Med Sci Monit 2014;20:706-10.
33. Joseph CA, Maroney MJ. Cysteine dioxygenase: structure and mechanism. Chem Commun 2007;32:3338-49.
34. Shi Y, Tokunaga O. Chlamydia pneumoniae and multiple infections in the aorta contribute to atherosclerosis. Pathol Int 2002;52(12):755-63.
35. Tufano A, Di Capua M, Coppola A, Conca P, Cimino E, Cerbone AM, et al. The infectious burden in atherothrombosis. Semin Thromb Hemost 2012;38(5):515-23.
36. Neumann FJ. Chlamydia pneumoniae-atherosclerosis link: a sound concept in search for clinical relevance. Circulation 2002;106(19):2414-6.
37. Rasmussen-Lathrop SJ, Koshiyama K, Phillips N, Stephens RS. Chlamydia-dependent biosynthesis of a heparan sulphate-like compound in eukaryotic cells. Cell Microbiol 2000;2(2):137-44.
38. Smolina K, Wright FL, Rayner M, Goldacre MJ. Determinants of the decline in mortality from acute myocardial infarction in England between 2002 and 2010: linked national database study. BMJ 2012;344:d8059.
39. Allender S, Scarborough P, O’Flaherty M, Capewell S. Patterns of coronary heart disease mortality over the 20th century in England and Wales: possible plateaus in the rate of decline. BMC Public Health 2008;8:148.
40. Roger VL. The heart failure epidemic. Int J Environ Res Public Health 2010;7(4):1807-30.
41. Braunwald E. Cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med 1997;337:1360-9.
42. Avila JL, Rojas M, Avila A. Cholesterol sulphate-reactive autoantibodies are specifically increased in chronic chagasic human patients. Clin Exp Immunol 1996;103(1):40-6.
43. Laranja FS, Dias E, Nobrega G, Miranda A. Chagas’ disease: a clinical, epidemiologic, and pathologic study. Circulation 1956;14:1035-60.
44. Murdoch JC, Rodger JC, Rao SS, Fletcher CD, Dunnigan MG. Down’s syndrome: an atheroma-free model? Br Med J 1977;2(6081):226-8.
45. Epstein CJ, Avraham KB, Lovett M, Smith S, Elroy-Stein O, Rotman G, et al. Transgenic mice with increased Cu/Zn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci USA 1987;84(22):8044-8.
46. Bittles AH, Bower C, Hussain R, Glasson EJ. The four ages of Down syndrome. Eur J Public Health 2007;17(2):221-5
47. Lai F, Williams RS. A prospective study of Alzheimer disease in Down syndrome. Arch Neurol 1989;46(8):849-53.
48. Brooksbank BWL, Balazs R. Superoxide dismutase, glutathione peroxidase and lipoperoxidation in Down’s syndrome fetal brain. Brain Res 1984;318(1):37-44.
49. Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol 1985;17(3);278-82.
50. Han X, Holtzman DM, McKeel DW Jr, Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J Neurochem 2002;82(4):809-18.
51. Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004;109(23 Suppl 1):III39-III43.
52. Jasińska M, Owczarek J, Orszulak-Michalak D. Statins: a new insight into their mechanisms of action and consequent pleiotropic effects. Pharmacol Rep 2007;59(5): 483-99.
53. Weitz-Schmidt G. Statins as anti-inflammatory agents. Trends Pharmacol Sci 2002;23(10):482-7.
54. Ridker PM, Lüscher TF. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J 2014;35(27):1782-91.
55. Maeda T, Kawane T, Horiuchi N. Statins augment vascular endothelial growth factor expression in osteoblastic cells via inhibition of protein prenylation. Endocrinology 2003;144(2):681-92.
56. Nakazato R, Gransar H, Berman DS, Cheng VY, Lin FY, Achenbach S, et al. Statins use and coronary artery plaque composition: results from the International Multicenter CONFIRM Registry. Atherosclerosis 2012;225(1):148-53.
57. Horwich TB, Hamilton MA, MacLellan WR, Fonarow GC. Low serum total cholesterol is associated with marked increase in mortality in advanced heart failure. J Card Fail 2002;8(4):216-24.
58. Rauchhaus M, Clark AL, Doehner W, Davos C, Bolger A, Sharma R, et al. The relationship between cholesterol and survival in patients with chronic heart failure. J Am Coll Cardiol 2003;42(11):1933-40.
59. Fonarow GC, Horwich TB. Cholesterol and mortality in heart failure: the bad gone good? J Am Coll Cardiol 2003;42(11):1941-1942.
60. Huxtable RJ. Physiological actions of taurine. Physiol Rev 1992;72(1):101-163.
61. Denger K, Laue H, Cook AM. Thiosulfate as a metabolic product: the bacterial fermentation of taurine. Arch Microbiol 1997;168(4):297-301.
62. Sturman JA, Hepner GW, Hofmann AF, Thomas PJ. Metabolism of [35S] taurine in man. J Nutr 1975;105(9):1206-14.
63. Milei J, Ferreira R, Llesuy S, Forcada P, Covarrubias J, Boveris A. Reduction of reperfusion injury with preoperative rapid intravenous infusion of taurine during myocardial revascularization. Am Heart J 1992;123(2):339-45.
64. Briet F, Keith M, Leong-Poi H, Kadakia A, Aba-Alkhail K, Giliberto JP, et al. Triple nutrient supplementation improves survival, infarct size and cardiac function following myocardial infarction in rats. Nutr Metab Cardiovasc Dis 2008;18(10):691-9.
65. Torres CL, Walker NJ, Rogers QR, Tablin F. Platelet taurine concentration can be predicted from whole blood taurine concentrations in dogs. J Nutr 2006;136(7):2055S-2057S.
66. Hayes KC, Pronczuk A, Addesa AE, Stephan ZF. Taurine modulates platelet aggregation in cats and humans. Am J Clin Nutr 1989;49(6):1211-6.
67. Crass MF 3rd, Song W, Lombardini JB. Cardiac muscle taurine: effects of acute left ventricular ischemia in the dog and anoxic perfusion of the rat heart. Recent Adv Stud Cardiac Struct Metab 1976;12:259-63.
68. Crass MF III, Lombardini JB. Loss of cardiac muscle taurine after acute left ventricular ischemia. Life Sci 1977;21(7):951-8.
69. Hasegawa T, Malle E, Farhood A, Jaeschke H. Generation of hypochlorite-modified proteins by neutrophils during ischemia-reperfusion injury in rat liver: attenuation by ischemic preconditioning. Am J Physiol Gastrointest Liver Physiol 2005;289:G760-G767.
70. Heslop CL, Frohlich JJ, Hill JS. Myeloperoxidase and C-reactive protein have combined utility for long-term prediction of cardiovascular mortality after coronary angiography. J Am Coll Cardiol 2010;55(11):1102-9.
71. Rosen H, Crowley JR, Heinecke JW. Human neutrophils use the myeloperoxidase-hydrogen peroxide-chloride system to chlorinate but not nitrate bacterial proteins during phagocytosis. J Biol Chem 2002;277:30463-8.
72. Ravnskov U, McCully KS. Review and hypothesis: vulnerable plaque formation from obstruction of vasa vasorum by homocysteinylated and oxidized lipoprotein aggregates complexed with microbial remnants and LDL autoantibodies. Ann Clin Lab Sci 2009;39(1):3-16.
73. McCarty MF. The reported clinical utility of taurine in ischemic disorders may reflect a down-regulation of neutrophil activation and adhesion. Med Hypotheses 1999;53(4):290-9.
74. Paasonen MK, Penttil a O, Himberg JJ, Solatunturi E. Platelet taurine in patients with arterial hypertension, myocardial failure or infarction. Acta Med Scand Suppl 1980;642:79-84.
75. Vogt W. Complement activation by myeloperoxidase products released from stimulated human polymorphonuclear leukocytes. Immunobiology 1996;195(3):334-46.
76. Schaffer SW, Jong CJ, Ito T, Azuma J. Effect of taurine on ischemia-reperfusion injury. Amino Acids 2014;46(1):21-30.
77. Yamori Y, Liu L, Ikeda K, Miura A, Mizushima S, Miki T, Nara Y on behalf of the WHOCardiovascular Disease and Alimentary Comparison (CARDIAC) Study Group. Distribution of twenty-four hour urinary taurine excretion and association with ischemic heart disease mortality in 24 populations of 16 countries: results from the WHO-CARDIAC study. Hypertens Res 2001;24(4):453-7.
78. Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 2008;294(2):C460-C466.
79. Chen Q, Paillard M, Gomez L, Li H, Hu Y, Lesnefsky EJ. Postconditioning modulates ischemia-damaged mitochondria during reperfusion. J Cardiovasc Pharmacol 2012;59(1):101-8.
80. Stipanuk MH, Ueki I. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis 2011;34(1):17-32.
81. McCully KS. Chemical pathology of homocysteine. V. Thioretinamide, thioretinaco, and cystathionine synthase function in degenerative diseases. Ann Clin Lab Sci 2011;41(4):301- 14.
82. Simon JA. Vitamin C and cardiovascular disease: a review. J Am Coll Nutr 1992;11(2):107-125.
83. Xu Y-J, Arneja AS, Tappia PS, Dhalla NS. The potential health benefits of taurine in cardiovascular disease. Exp Clin Cardiol 2008;13(2):57-65.
84. Liao X-B, Zhou X-M, Li J-M, Yang J-F, Tan Z-P, Hu Z-W, et al. Taurine inhibits osteoblastic differentiation of vascular smooth muscle cells via the ERK pathway. Amino Acids 2008;34(4):525-30.
85. Kramer JH, Chovan JP, Schaffer SW. Effect of taurine on calcium paradox and ischemic heart failure. Am J Physiol 1981;240(2):H238-H246.
86. Kingston R, Kelly CJ, Murray P. The therapeutic role of taurine in ischaemia-reperfusion injury. Curr Pharm Des 2004;10(19):2401-10.
87. Olszowski S, Olszowska E, Kusior D, Szneler E. Sulphoacetaldehyde as a product of taurine chloramine peroxidation at site of inflammation. Amino Acids 2002;22(2):145-153.
88. Guo W, Li JY, King H, Locke FB. Diet and blood nutrient correlations with ischemic heart, hypertensive heart, and stroke mortality in China. Asia Pac J Public Health 1992;6(4):200-9.
89. Rahman K, Lowe GM. Garlic and cardiovascular disease: a critical review. J Nutr 2006;136(3):736S-740S.
90. Wollin SD, Jones PJH. α-Lipoic acid and cardiovascular disease. J Nutr 2003;133(11):3327-30.
91. Bilska A, Dudek M, Iciek M, Kwiecień I, Sokołowska-Jezewicz M, Filipek B, et al. Biological actions of lipoic acid associated with sulfane sulfur metabolism. Pharmacol Rep 2008;60(2):225-32.
92. Chou MM, Vergnolle N, McDougall JJ, Wallace JL, Marty S, Teskey V, et al. Effects of chondroitin and glucosamine sulfate in a dietary bar formulation on inflammation, interleukin-1β, matrix metalloprotease-9, and cartilage damage in arthritis. Exp Biol Med (Maywood) 2005;230(4):255-62.
93. Bhuiyan MS, Takada Y, Shioda N, Moriguchi S, Kasahara J, Fukunaga K. Cardioprotective effect of vanadyl sulfate on ischemia/reperfusion-induced injury in rat heart in vivo is mediated by activation of protein kinase B and induction of FLICE-inhibitory protein. Cardiovasc Ther 2008;26(1):10-23.
94. Sochman J. N-acetylcysteine in acute cardiology: 10 years later: what do we know and what would we like to know?! J Am Coll Cardiol 2002;39(9):1422-8.
95. Woods KL, Fletcherk S, Roffe C, Haider Y. Intravenous magnesium sulphate in suspected acute myocardial infarction: results of the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2). Lancet 1992;339(8809):1553-8.
96. Del Giudice E, Tedeschi A, Vitiello G, Voeikov V. Coherent structures in liquid water close to hydrophilic surfaces. J Phys Conf Ser 2013;442(1):012028.
97. Pollack G. Cells, Gels and the Engines of Life: A New, Unifying Approach to Cell Function. Seattle, WA: Ebner and Sons, 2001.
98. Pollack G. The Fourth Phase of Water: Beyond Solid, Liquid, and Vapor. Seattle, WA: Ebner and Sons, 2013.
99. Trivedi DP, Hallock KJ, Bergethon PR. Electric fields caused by blood flow modulate vascular endothelial electrophysiology and nitric oxide production. Bioelectromagnetics 2013;34(1):22-30.
This article appeared in Wise Traditions in Food, Farming and the Healing Arts, the quarterly magazine of the Weston A. Price Foundation, Summer 2017.
🖨️ Print post
Your article “Cholesterol sulfate deficiency and coronary heart disease” was of great interest.
I have read that it is the ultra-violet B rays (UVB) on the skin that synthesize vitamin D3 and that the sun must be above an angle of 50 degrees in order for the UVB to penetrate the earth’s atmosphere. It appears that it is not possible to get vitamin D3 from the sun in most of the country during the 4 to 6 months a year when the angle of the sun does not get to 50 degrees. Does the synthesis of cholesterol sulfate also depend on UVB rays?
Another article that states that most of the year vitamin D3 requirements from sun can be obtained in a matter of minutes by sun exposure at the noon hour. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3257661
I have avoided the sun for years on the in part because of skin cancer problems and because I assumed that I did not need sun exposure since I was taking vitamin D3 supplements. I walk a couple of miles a day in the morning but don’t get any UVB rays because of the low sun angle. I plan to try getting a few minutes sun at the noon hour hopefully to get some cholesterol sulfate synthesis.
You recommended eating sulfate-containing foods such as seafood, eggs, cheese, grass-fed beef, cruciferous vegetables, garlic and onions. I assume milk would also have sulfate since cheese is made from milk. It seems to me that because all the animal foods listed contain both cholesterol and sulfur, they would also contain ready made cholesterol sulfate, especially those foods from grass fed animals that get a lot of sun exposure. My diet includes a good bit of fermented raw milk and raw milk cheese from grass fed cows and eggs from grass fed chickens which I like to think provide some ready made cholesterol sulfate. Does that sound reasonable?
Thank you for your wonderful article. It has given me a lot to think about.
Being in the 27% of people who have had some sort of (near fatal) cardio vascular event and no conventional risk factors (e.g. smoking, overweight), I’m interested in exploring alternative hypotheses such as Stephanie Seneff’s chondroitin sulfate (CS) deficiency.
But I can’t find out how to test for CS deficiency. The above article suggests the inflammatory marker of interluekin 1-beta.
Elsewhere, the following have been suggested:
– High total cholesterol (but that could be familial hypercholesterolaemia)
– Coronary arterial calcium score (but that could be vit K2 problems)
– High homocysteine (I thought that was a vit B deficiency)
– High lp(a) (I thought that was vit C deficiency)
– Vit D3 (if that it is low, does it follow that CS is low.)
– lp-pla 2 (no idea what this is).
Natural sunlight, diet (e.g. bone broth and garlic) and bath salts would appear to be the solution. However, what supplements are recommended? Specifically:
You can buy CS supplements. But I’ve read that methyl sulfonyl methane (MSM) has been recommended. Which is best? And why can’t the vit D3 supplement manufacturers add CS to their vit D3 supplement to get the ultimate sunshine supplement?
Can anyone help on my CS testing and supplement query?
Thanks
I have followed Dr Seneff for several years. Did you know that wheat fields are sprayed with glyphosate 10 days before harvest to expedite combining? Sulfur can be easily taken as MSM and I have taken a tsp./day for 2-3 years on morning coffee. A pound jar of powder cosst about $14 from Amazon. The head of the dematology departmentat my medical school, the U of MN (45 deg. N. latitude) applies sunscreen even in the winter. I’ll have to check and see if he is still alive as I listened to his lecture about 10 years ago. At this time I am going with the theory that it is INSULIN resistance and HYPERINSULINEMIA that is the injurious agent that causes ulceration of the endothelium. There is no doubt some pressure/flow mechanical factors too as some sights are typically more at risk —bifircations of the carotids and femoral arteries.
Amazing, thank you! I have three friends well into their 80’s and doing great. Two of the three had serious health issues in their 60’s. Both had heart failure, one had some form of cancer and the other had fibromyalgia. They both lost their husbands about the same time and started outdoor activities such as walking and golf. They both recovered from their issues and have done well for more than 20 years. (could have been the husbands’) The third friend has always had jobs that had him walking outdoors. He is doing great in his mid 80’s. He says he lost both his parent (one to heart disease and one to cancer in their 50’s and 60’s. Lost all his siblings at similar ages. Apparently the only thing that none of them had in common with him was sun exposure daily.
see: http://www.practicingmedicinewithoutalicense.com
heart disease cure in action
Very interesting article. How does ApoE3 vs ApoE4 allele affect the above? Would the advice be the same regarding eating cheese? I’ve heard people with ApoE4 allele really crave cheese, but should not eat it because of the saturated fat. Do they crave cheese because they need the sulfur? Is cheese really bad for people with ApoE4?
i’ve gone vegan for one and a half years, then suffered a heart attack, had a stent put in, and had a pulmonary embulism detected the next day. this article is very lovingly important to me.
I am only 52, and lost 60 lbs (240 to 180) but still this happened to me. I was eating allot of roundup products!!!
thankyou so much, and i look forward to your new book comming out.
I have been researching (I’m not a doctor) the misguided medical policies and research around cholesterol for over 15 years. Much of Ancel Keys hypothesis was flawed and the subsequent vilification of cholesterol has led to the overuse of statins. Mind you, the pharma industry makes billions of dollars from the proliferation of statins. One may suggest that this is why so many doctors and researchers who have spoken out against statins have been sued, threaten with cuts to funding and otherwise fired from research institutes. Probably the single biggest threat to the cardio-vascular system and the buildup of plaque is smoking. Obviously, other environmental toxins may be significant as well. Animal fat, eggs, cheese and other foods simply are not the culprits.
There is evidence (successfully buried by big pharma) to indicate that those with high levels of LDL will live longer. Interestingly, the male members of my in-laws who have all been diagnosed with “dangerously high cholesterol (LDL)” all live well into their 90’s with no cardiovascular disease or statin consumption.
I am trying to figure out why Zetia (or its generic) is added to statins if it doesn’t help your heart. I would love to know because I never wanted to take statins and now the medical establishments seems to want me to take both. Any thoughts?